Transthyretin (TTR) amyloidosis (ATTR) is a rare, underdiagnosed, and life-threatening disease with limited treatment options that can damage the heart and/or nervous system. BridgeBio’s mission is to improve the morbidity and mortality of patients with ATTR through the discovery and development of a novel therapeutic through its affiliate, Eidos Therapeutics.
Our Research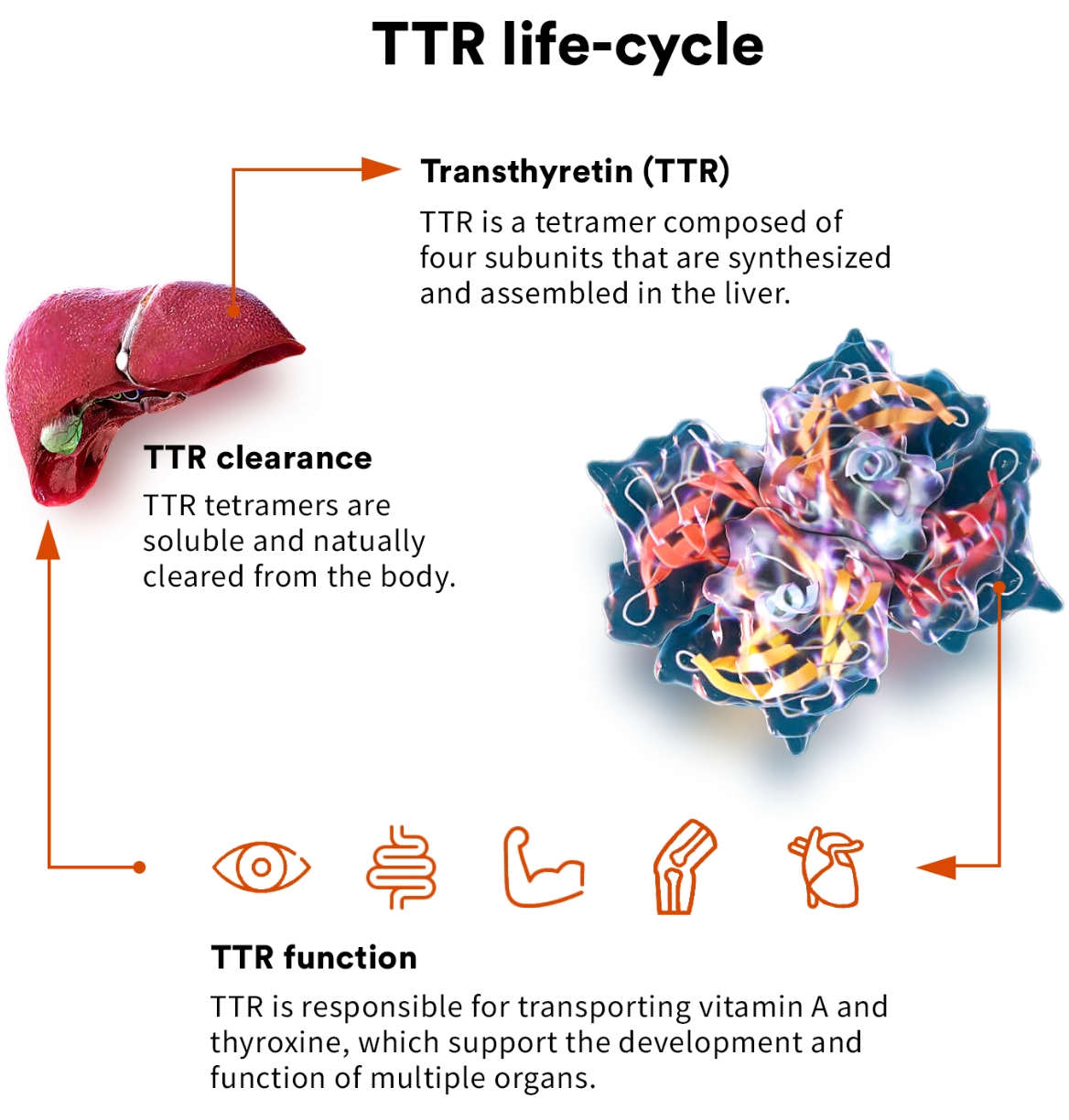
transthyretin (TTR)
TTR is a highly evolutionarily conserved protein that has been found to circulate in the blood of all vertebrates. As a circulating protein primarily made in the liver, TTR plays a key role in the transport of thyroxine and vitamin A and is thought to have a role in preserving cognitive function.1,2
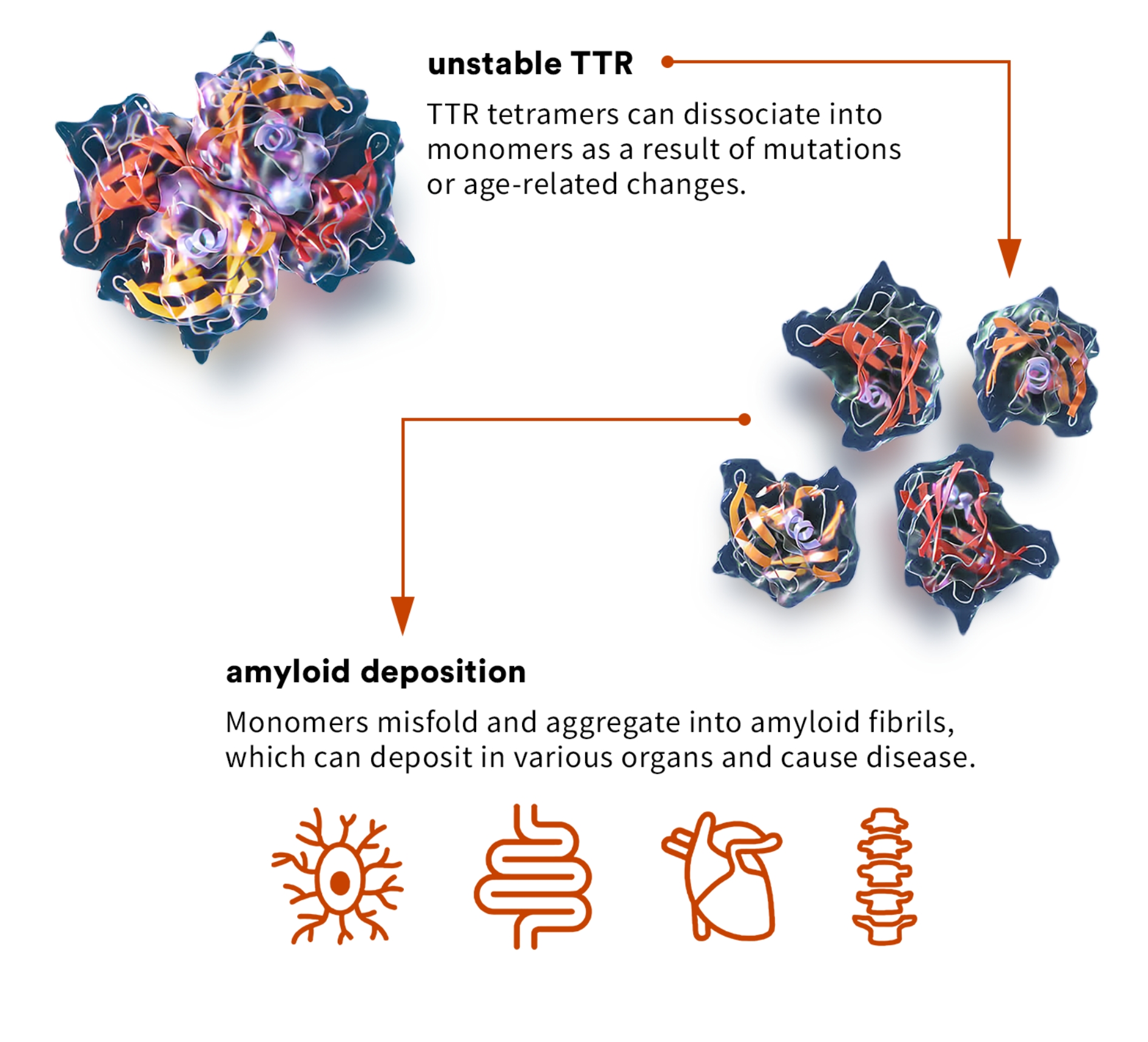
transthyretin amyloidosis (ATTR)
ATTR cardiomyopathy, or ATTR-CM, is a form of amyloidosis caused by destabilization of TTR and subsequent accumulation of misfolded TTR protein in the myocardium. Approximately 13 to 19% of all patients with heart failure with preserved ejection fraction, commonly referred to as HFpEF, may have ATTR-CM, though it is currently significantly underdiagnosed.3 ATTR-CM often dramatically impairs the quality of life, functional independence, and life expectancy of patients, as well as impacting caregivers due to the progressive nature of the disease. If left untreated, life expectancy from diagnosis is approximately four years.
ATTR can be caused by genetic variants, which can be associated with either destabilization or increased stabilization. Science has found:
- In more than 130 destabilizing ATTR-causing variants, the severity of disease is associated with the level of destabilization caused by each variant.
- Individuals who inherit stabilizing variants like T119M are partially or completely protected against the development of ATTR.
related news
References
- Liz MA, et al. Neurol Ther. 2020;9(2):395-402.
- Vieira M, Saraiva MJ. Biomol Concepts. 2014;5(1):45-54.
- AbouEzzeddine OF, Davies DR, Scott CG, et al. Prevalence of Transthyretin Amyloid Cardiomyopathy in Heart Failure with Preserved Ejection Fraction. JAMA Cardiol. 2021;6(11):1267-1274. doi:10.1001/jamacardio.2021.3070